《從天堂墜入地獄,我爬回人間,一位罕見病兒童爸爸的心路歷程》
2020-10-13
轉載自澎湃新聞(2020-10-10)
作者:慢飛天使 果殼病人
花卷剛出生的時候,我擔心孩子遺傳了我的高度近視,覺得小小年紀就戴眼鏡是天大的事。
後來,花卷被評價為發育遲緩,我擔心的是孩子可能會笨一點、慢一點,需要家長更多的付出。再後來,核磁檢查出先天性腦發育不良,我只希望她長大後能自理。
最後,基因檢測確定病因,FOXG1基因突變導致雷特綜合徵,我唯一的希望就是她能順利長大。
對於有的人來說,活著,真的就已經竭盡了全力。
孩子追物不太好
我是一個普普通通的小鎮青年,跟多數人一樣上學、就業、結婚、生子。也許是我的前半生過得太平淡了,老天爺想要給我點刺激,2018年一切都變了。
2018年9月26日,北京的夏天還是那麼悶熱,花卷媽媽的一個電話打斷了我的工作,“什麼時候帶花卷去做個系統檢查,她情況不太好”。我愣了一下問:“什麼不太好?”在我眼裡,6個月大的花卷能吃能睡、白白胖胖,和其他孩子沒什麼不一樣。
花卷媽媽說,今天去做常規體檢,花卷的追物不太好,本以為是視力有點問題,但大夫提了一句可能跟腦子發育有關,讓去做做檢查。
幾分鐘就斷定發育有問題,還要測基因?
我工作的地方離首都兒科研究所比較近,就先帶花卷去了兒研所看病。
那裡的患者永遠都是那麼多,一位年紀很大的醫生大致聽了來檢查的原因,摸了摸孩子的肌張力,用手測了一下頭圍,就開始一邊開單子一邊講:“孩子的肌張力有點異常,而且頭圍偏小,做一下全面的發育評估吧,發育肯定是有問題的,常規檢查不一定能查出原因,建議你們做個基因檢測。”
整個就診過程不超過5分鐘,有3分鐘是在開單子。直到從診室出來我都沒反應過來,這就看完了?哪跟哪都沒說就有問題了?還要測基因?
進診室之前我篤定花卷是沒問題的,就是想讓醫生看一眼,明確說孩子沒有問題,讓我們心裡踏實下來。沒想到結果跟預想的完全不一樣,我一下子就慌了。
兒研所名聲顯赫,這裡的大夫做出的診斷其實是很有權威性的,但是這麼幾分鐘就要讓我接受花卷有問題的事實,對於我來說太突然了。於是跟家里人商量了一下,決定換個地方再看看。
現在回想,這個決定很難說是對是錯,如果當時就在兒研所直接做了基因檢測,有可能會省掉後面很多的折騰,只不過是鴕鳥心理作祟,不想面對現實。
檢查還沒做完,就開始了康復治療
我們帶著花卷從兒研所出來,直奔北京市兒童醫院。又是一系列的辦卡、掛號、就診,這次醫生更仔細地詢問了孩子吃飯、睡覺的各種情況,做了一些基本的能力測試,也大致檢查了下手腳肌張力。
最終的結論是花卷有些發育遲緩,但是很多表現都介於正常和異常之間,還是需要先做系統的發育評估。
等待評估的時候,我一直想著自己算個好人,開車從來不和行人搶道,地鐵上遇到不方便的人肯定主動讓座,遇到舉手之勞的小事都會主動伸手幫一把。都說好人有好報,我應該不會這麼倒霉吧。
系統評估其實就是對孩子發育情況的更詳細檢查,結果出得很快,一共20項發育指標,花卷有7項不合格,包括大運動、精細運動、適應能力、語音能力和社交行為。
拿到報告單的時候,我的心涼了一半,這等於是坐實了發育遲緩。不過發育遲緩只是表象,6%~8%新生兒有這個表現,其中只有不到10%是由各種病變引起的,剩餘90%屬於正常現象,不需要特殊治療。
具體是什麼原因導致花捲發育遲緩,還要做更專業的檢測來判斷,包括微量元素、遺傳代謝和頭部的核磁檢查,醫生建議在結果沒出來之前就開始康復治療。
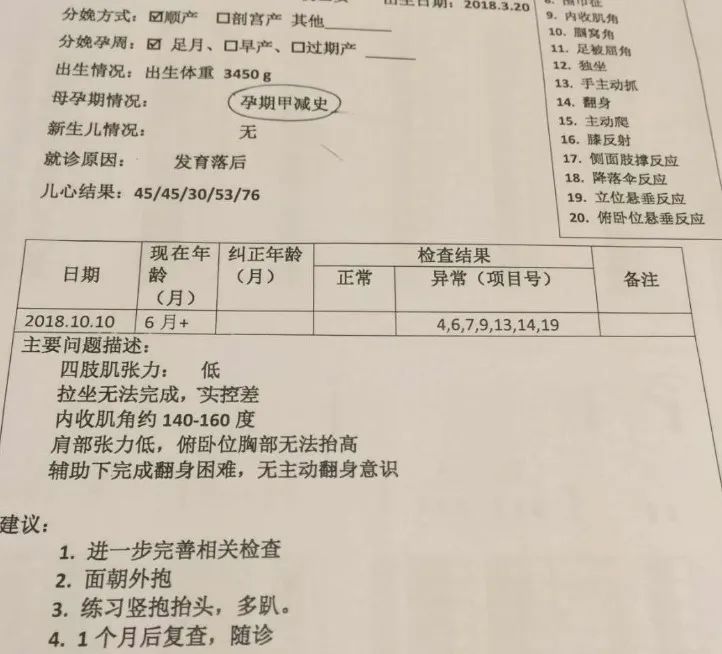
發育評估報告結果 | 作者供圖
就這樣,我們開始了一邊康復一邊檢查的日子。微量元素和遺傳代謝的結果都正常,我心裡也越來越踏實,沒準花卷就只是單純的發育遲緩,說話慢一點、走路慢一點,沒關係,我們可以慢慢來。
一直到2個月之後,我拿到頭部核磁的檢查結果“大腦半球腦迴路略簡單,雙額葉腦溝增深,胼胝體前部薄,綜合考慮腦發育不良”。 12月的北京,我坐在兒童醫院的樓梯上,汗完全打濕了內衣,一滴一滴地順著臉頰滴到地上。當時大腦完全已經空白了,只有一個想法不停迴盪:怎麼會這樣。
不管是什麼原因導致了發育遲緩,都沒有任何藥物可以治療,只有物理療法,通過日復一日的鍛煉促進患兒發育,增強大腦正常部分的代償功能,替代發育不良的部分,才能改善症狀。諮詢了北京兒童醫院的醫生之後,我們開始了漫長的康復生涯。物理治療、職能治療、言語治療、認知,每天的課程排得滿滿噹噹,偶爾不去醫院康復的時候,我們也會在家鋪上地墊,讓花卷在上面鍛煉。
日子一天一天過去,花卷也在“龜速”進步著,慢慢可以自己翻身,只是好像沒有翻身的興趣;慢慢可以從地墊的一頭“咕丘”到另一頭;慢慢也能坐得越來越直。很多時候我認命地想,也就這樣了,我和花卷媽媽都是急性子,結果有了個什麼事都比別人“慢三拍”的孩子,也挺好的。
一頓拳打腳踢後,生活還要給你補上一棍子
但生活往往就是這樣,對你一頓拳打腳踢,在你服了之後還要再來上一棍子。半年之後,花卷有了新的症狀,半夜睡著覺會突然痙攣,手腳緊繃、腦袋往前伸、眼睛無意識地上翻,一直到哭出來才能緩解。輕的時候一兩分鐘就好了,重的時候要持續十幾分鐘,甚至臉憋得發黑、嘴唇發青。
腦發育不良的孩子經常會伴有癲癇發作,而每次發作都會導致發育倒退。我們不敢耽誤,又一次踏上了求診的道路,朋友說北京大學第一醫院婦產兒童醫院的神經內科專家很厲害,於是這次我們選擇了北大婦兒。
醫生問過常規問題,又加了一句“孩子一直搓手、絞手是從什麼時候開始的”。看著我一頭霧水的樣子,醫生解釋道,無意識的手部刻板動作是一個罕見病---雷特綜合徵的典型症狀之一,建議我們去掛另一位包醫生的號,她是國內研究這個病最好的醫生之一。
包醫生看過花卷,說從臨床上看確實存在雷特綜合徵以及由其引起癲癇的可能性,但還是要以腦電圖和基因檢測的報告為準。腦電圖的報告很快就出來了,按照包醫生的話說,“我從來沒有見過腦發育不良的孩子有這麼正常的腦電圖”,也暫時排除了癲癇。那段時間花卷痙攣的程度及頻率也在減輕,包醫生就建議繼續觀察一段時間。
基因檢測本來就耗時,又趕上新冠疫情,過了4個月我才拿到報告。等待期間我上網查了雷特綜合徵,隱隱約約覺得花卷有很多症狀能對得上,已經有了心理準備,但是看到“FOXG1基因突變導致先天性雷特綜合徵”這幾個字,我還是完全懵了。
當時體驗到了書上總看到的一句話“感覺心一直不停地往下掉”,真的覺得心臟一直不停地下落,卻總也落不到底。

基因檢測報告 | 作者供圖
不過調整好心態之後,一家人反而看開了很多,或者說句不恰當的話,我們認命了很多。這次終於知道了最根本的病因,為什麼花卷會發育遲緩;為什麼別的孩子通過康復都有肉眼可見的進步,只有她進步緩慢;為什麼她從小入睡困難,到現在還不會走、不會爬、不會說話,這些問題都有了答案。
雷特綜合徵主要是三個基因位點出現問題導致的,其中絕大多數都是MECP2基因突變,另外兩個基因FOXG1和CDKL1導致的被稱為非典型或者亞型雷特綜合徵,現在世界上已知的後兩種基因導致的病例一共不到1000例,真是小概率中的小概率事件。
由於這個病的發病率實在太低了,現在還沒有對症的藥物,而且花卷的基因突變類型屬於罕見病中的少數派,藥物治療更是遙遙無期。
但是無藥可治並不等於無路可走,類似的基因疾病比如脊髓性肌萎縮症,現在已經有靶向藥上市而且有很好的療效。
只是眼下基因治療費用動輒一年上百萬,而且部分罕見病的診斷和治療技術,國內只有寥寥幾個發達地區具備,在三四線城市及偏遠地區,別說治療,有更多的人可能直到最後都沒有搞明白病因是什麼。
但行好事,莫問前程
花卷確診之後,我們加入了雷特綜合徵和單獨的FOXG1基因突變的病友群,家長們在群裡互相打氣,分享孩子康復方面的經驗。
我們還通過網絡了解到,一對韓國博士夫婦也有一個FOXG1基因突變的孩子,他們的團隊正在美國布法羅大學進行FOXG1基因的研究,目前已經進行到小鼠實驗的階段。我們也希望聯繫到國外的專業團隊,更好地促進國內這種罕見疾病診斷、治療技術的發展,幫助更多像我們一樣,或者某些方面還不如我們的人群。

韓國的李洙京博士、李載博士與他們患雷特綜合徵的大女兒在一起 | foxg1research
現在,受疫情影響而中斷的康復治療慢慢恢復了正常,花卷又開始了每天不間斷的訓練。
我很感謝花卷媽媽,本來覺得這個90後的女孩自己都是個孩子,但她在經歷了這樣的打擊後瞬間成長。由於我還要正常上班,花卷媽媽全職在家24小時陪護花卷,相信她面臨的精神壓力只會比我更大。不過她仍然滿懷希望,堅定地認為花卷會越來越好,與我相互鼓勵、互相扶持。
罕見病就像一個巨浪打破了原本平靜的水面,有的家庭傷痕累累地抗了過去,有的家庭被拍打得支離破碎。
從發現花卷有問題到確診,經歷了將近一年半的時間,這個過程中的種種辛苦很難用言語來描述。這期間我流的淚超過了之前三十多年的總和,小時候調皮,被父親不小心打得胳膊骨裂都沒有掉一滴眼淚的我,被電影《我不是藥神》中的情節感動得淚流滿面。
花卷現在還小,我們還能照顧,不敢想四五十年後,等我和孩子媽媽老了、不在人間了,她要如何生存。可能眼前我們能做的就是好好康復,盡量不要讓花卷的發育落下太多,然後努力工作攢錢,期待醫學發展到那一步的時候我們有足夠的經濟能力。
最近幾年有句話特別火,叫做“不忘初心,方得始終”,這句話很好,但是相比之下,我更喜歡這句話的前半句:
但行好事,莫問前程。
醫生點評
章清萍,包新華 | 北京大學第一醫院兒科醫師
由於雷特綜合徵比較少見,並且在發病初期症狀不典型,因而患兒的診斷常常像本文提到的那樣經歷波折。當患兒出現發育遲滯甚至倒退、頭圍增長緩慢,以及手部刻板動作時,需警惕雷特綜合徵的可能。
雷特綜合徵是女性重度智力低下的主要原因之一,新生女嬰的發病率為1/10000。典型雷特綜合徵表現為出生後6~18個月生長發育正常(少數早期出現發育遲滯),隨後出現發育停滯或倒退,喪失已獲得的技能,如手功能及語言等,出現手刻板動作,包括搓手、絞手、吃手及揪頭髮或衣物等;多伴有孤獨症樣行為及痛覺異常。
不典型雷特綜合徵包括Hanefeld型雷特綜合徵(也稱為早發癲癇型雷特綜合徵)和先天型雷特綜合徵。 Hanefeld型雷特綜合徵患者多在生後5月內出現癲癇發作,為難治性癲癇;智能及運動發育嚴重落後;除具有早發性癲癇腦病的特徵外,還具有以上所述手刻板動作。
先天型雷特綜合徵的臨床特點,包括生後即表現為全面發育異常,生後4月內出現嚴重小頭;生後5月內出現倒退,呈嚴重智力運動遲滯,不能行走;具有典型雷特綜合徵的自主神經功能異常的表現,如手腳小且涼、外周血管運動異常、睡眠障礙、清醒時呼吸節律異常。
雷特綜合徵為單基因遺傳性疾病,遺傳方式包括X連鎖顯性遺傳及常染色體顯性遺傳。甲基化CpG結合蛋白2基因(MECP2)為典型雷特綜合徵的主要致病基因,Hanefeld型雷特綜合徵的主要致病基因為MECP2和細胞週期蛋白依賴激酶樣5(CDKL5)基因,先天型雷特綜合徵的主要致病基因為MECP2及FOXG1基因。
MECP2及CDKL5基因位於X染色體,FOXG1基因位於14號染色體。遺傳性疾病傳統意義上為“垂直”傳遞,即父母遺傳給子代;亦有父母無基因突變,患兒自身出現突變,即為新生突變。如患兒致病基因明確,建議其父母完善基因檢測,必要時完善生殖細胞嵌合突變分析;如父母體細胞及生殖細胞均含有突變,則遺傳給子女的概率為50%;如單純生殖細胞嵌合突變,亦有一定概率遺傳給下一代。因而,高危家庭再次孕育時建議完善產前諮詢及產前診斷。
雷特綜合徵在世界各國均有報導,目前我國已診斷並隨訪患兒上千例。疾病診斷主要依靠臨床表現,基因檢測可以協助診斷。
同文中所述,雷特綜合徵目前無特異性的治療方法,以對症支持治療為主,如針對癲癇、脊柱側彎及肺炎等治療。而康復訓練貫穿疾病的始終,以維持、改善現有功能,減緩倒退。
自1986年至今,已完成並報導的針對雷特綜合徵的臨床治療研究有數十項,目前仍在探索中。雷特綜合徵在基因水平的治療研究仍處於細胞實驗階段,尚處於摸索階段,但未來隨著基因編輯技術的不斷發展,以及發病機制的揭示,基因治療有可能成為一種重要的治療手段。
個人經歷分享不構成診療建議,不能取代醫生對特定患者的個體化判斷,如有就診需要請前往正規醫院。